The code that I’ve been working on for a while to compute excited state absorption spectra can be found here https://gitlab.com/plusxmartin/nwproj , the wiki page contains more details. This is work in progress. For more info please contact me
Germanium Fluorides
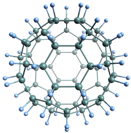
Our recent work on Germanium Fluoride nanocages was accepted by the Journal of the American Chemical Society. In this work, we studied with DFT a series of high-symmetry molecules with Formula GenFn, where n is between six and sixty. The molecules we studied show optical transparency or semi-transparency, electron delocalization, LUMO energies below -4 eV, and gaps in the range 1.6-3.2 eV. Although the formation energies are high, the fullerene-like systems have isomers with lower ground-state energies, where some Fluorine atoms are located inside the cages. So the fullerene below could undergo a transformation with F atoms preferring to be inside; but the barrier for such a transition should be very high. Link https://pubs.acs.org/doi/10.1021/jacs.8b11259
Releasing space in a MacBook Pro
Many of the applications I use require the gigantic package Xcode, which is often updated. An issue with these updates is that they always re-install a set of platforms. These are used I think to simulate several devices. Unfortunately they take up a lot of space. The platforms are located in /Applications/Xcode.app/Contents/Developer/Platforms. `du -hd 1` shows
1.4G ./AppleTVOS.platform
2.2G ./iPhoneOS.platform
1.1G ./WatchOS.platform
46M ./MacOSX.platform
5.9M ./WatchSimulator.platform
4.5M ./AppleTVSimulator.platform
6.3M ./iPhoneSimulator.platform
4.7G
I don’t use any of these except MacOSX (I tried to delete it the other day and learned it’s actually required by Xcode). So erasing them (except again MacOSX.platform) saves about 4.7GBs in space, much needed in a laptop with only 128GBs in SSD 🙁
Papers on Locally Coupled Open Subsystems
Back in July (2018), our paper on molecular dissociation, size consistency, and fractional charges, was published in the Journal of Chemical Physics. https://aip.scitation.org/doi/pdf/10.1063/1.5038557
This work introduces our new family of theories, locally coupled open subsystems (LCOS), which is initially intended as a set of electronic structure methods that can lead to controllability of the computational scaling of quantum chemical calculations. A common problem with standard DFT calculations is that electrons/holes usually are delocalized when a single molecule (or more) interacts with a surface that is subject to a bias. A way to resolve this problem is to couple the molecule to the surface in such a way that one can perform separate DFT calculations on the molecule and surface, and then one can introduce another DFT method for the coupling, so the fragments exchange energy and electron charge. This is advantageous from fundamental and computational perspectives. The theory is flexible, and has a good number of potential applications in chemistry and physics.
In our most recent paper, https://journals.aps.org/pra/abstract/10.1103/PhysRevA.98.062505, the full formalism is described. This work includes the possibility of using correlated wave function methods instead of DFT to describe the electronic structure of the molecule and the surface, for instance. The paper also presents the formalism to ensure the spin symmetry of the whole system is preserved, and a way to perform time-dependent propagation of the state of the entire system: The electronic occupations of the fragments can now be obtained as a function of time, meaning that we can let the nuclei evolve as well and/or apply an external field and track the flow of energy and electrons. An important point of the LCOS work is that the computational implementation is simple. The DFT version of LCOS, for example, uses local potentials and density functionals in such a way that computing the charge and energy transferred between the molecule and the surface is not difficult to obtain; LCOS in this context uses ensemble DFT, which features the needed derivative discontinuities!. The next step I am working on is centered on implementing LCOS and applying it to problems where common DFT methods have trouble. The implementation (within NWChem) mostly consists of managing pieces of established code, and using these pieces to make (models of) molecules interact accordingly to the theory. (Update 01/30/19: It’s cold in Chicago!, -20F. Although this is optional, I found several ways to compute the lambda value, or matrix. The implementation will also help in this direction. Update 10/04/19: manuscript under preparation)
Updating the cookbook
Update 05/26, added an incomplete draft of new chapter, yay😀, there’s still a lot of work to do. Update 05/19, found some nasty typos in the old document.
04/22. For a while I’ve been wanting to expand the cookbook, with applications. The theory part will always be incomplete [although it really needs chapter(s) on strong correlation, molecular dissociation, and electronic transport], advances happen by the hour in the literature. But computational applications are simply too important. So I hope the cookbook will double in size this year, with the new half devoted to applications. A crazy winter quarter is over!, and spring is slowly warming up, so it’s time to add recipes to the notes
ACS Energy Letters paper out

Check out our ‘just accepted‘ paper [at ACS Energy Letters] on future prospects in quantum chemistry for research in materials and energy research (link here). Given the fast paced advancements in artificial intelligence and learning algorithms, we believe they will have considerable impact in computational chemistry. If the application of DFT survives this wave, an algorithm may choose or create a dft model for you to study some specific material, and its properties; or even come up with a candidate material for a given application. Otherwise, new forms of computer-generated theoretical models might arise for these purposes.
Book on Group Theory
Browsing for irreps the other day I found this recent book on group theory and applications. It’s an interesting and useful reference, includes formalism and examples of groups in solids and molecules. Link:
Printing correlation energy in nwchem dft calculations
I learned the hard way (examining the code, the variables nexc [sometimes nExc] and idecomp are confusing) that the keyword “decomp” activates decomposition of the exchange-correlation energy into exchange and correlation. So the dft block should look like:
dft
mult
xc…
decomp
end
Inexpensive Stabilization of a Perovskite Solar Cell
A news article popped up in my newsfeed about a record in stability. This article shows that copper thyocianate can work as hole-transport material in a lead-based perovskite solar cell. They seem to achieve 1k-hour stability. Very promising indeed. Does lead have its days numbered?
JCP paper out
Some of my work on the Runge-Gross theorem (the Hohenberg-Kohn theorem of TDDFT) was published this morning (JCP, 147, 134110). This theorem states that the time-dependent electronic density determines the potential uniquely (up to a constant), so we can define observables as time-dependent density-functionals. This manuscript extends the theorem to include time-dependent densities and potentials that are not Taylor expandable with respect to time. The Supp Info includes an alternative method, and an application to time-dependent current-density functional theory.