Congrats to our collaborators from the Mock lab for a nice report on light-induced dinitrogen activation. Starting with a [POCO]Cr(Benzyl) complex, this work shows that light can cleave the benzyl-Cr bond, leading to the formation of a N2 bridged Cr dimer complex. Link
Splitting Molecules
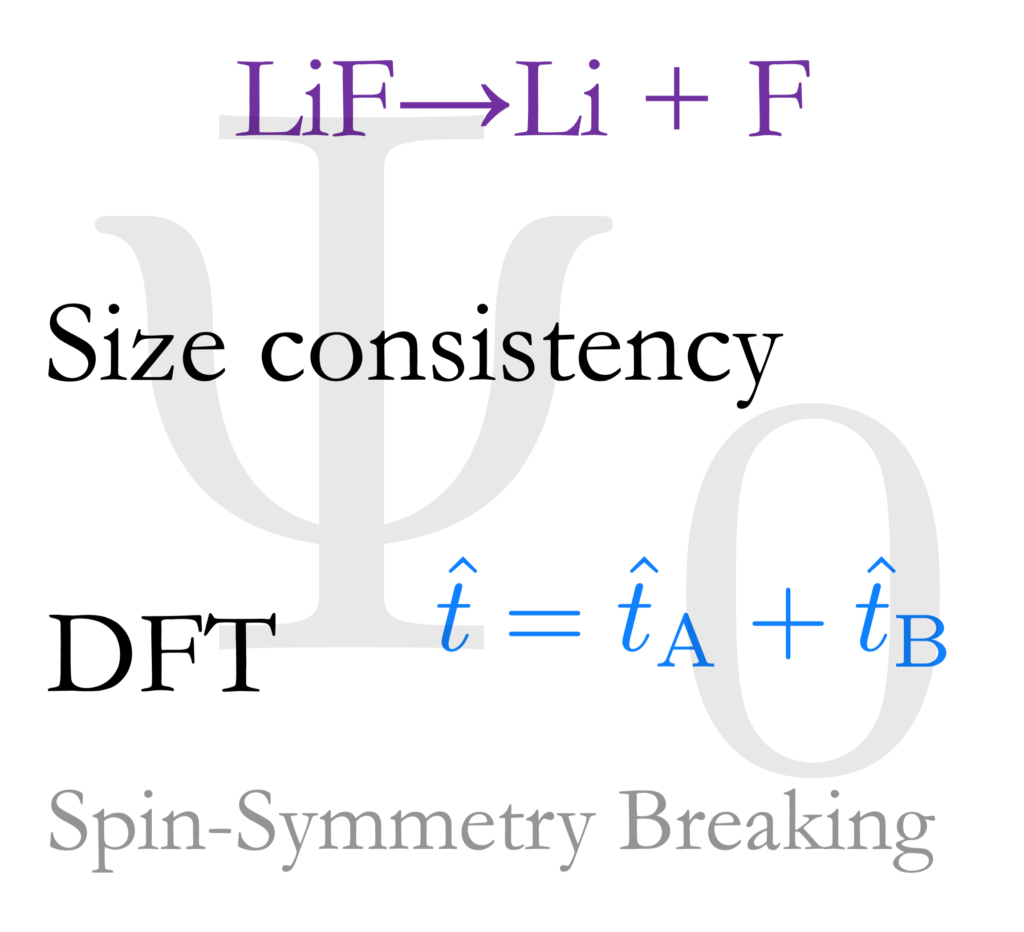
Following up our previous work that uses an idea from coupled cluster but in DFT calculations, we reported further application of this concept to compute the binding energies of diatomic molecules and to illustrate the elimination of the well-known problem of fractional charges. This work is the result of productive efforts by Greta Jacobson (2021 REU student at the time, now at U Washington Chemistry), Dr. Marmolejo-Tejada, and myself. DFT simulations struggle sometimes to predict properties of systems where one or more bonds are “stretched” significantly in such a way that fragments with unpaired electrons emerge. Our work shows a practical way to avoid these problematic charges. In this paper we also discuss procedures that save computational resources for future applications. Big thanks to the #LatinXChem 2021 organizers and ChemPhysChem for their invitation to contribute to this special issue in Latin American Physical Chemistry. Link to publication.
Propagating Arbitrary Quantum States Within Coupled-Cluster Theory
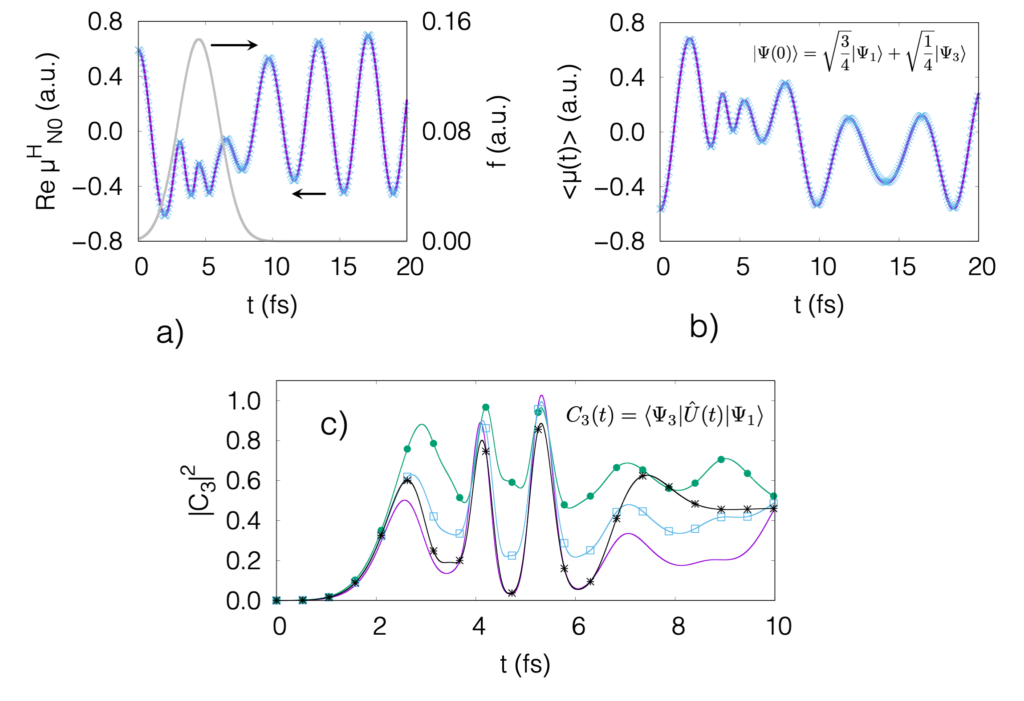
This paper published in Physical Review A shows our theory based on coupled cluster to propagate in principle arbitrary superpositions of quantum states, while maintaining the desired property of size-extensivity. It, size-extensivity, is a fundamental property that physical “large” quantum systems have, and that is difficult to satisfy theoretically speaking. The idea is based on the concept of “second response”: If a system that is initially a linear combination of ground and excited state is propagated, where the “weight” of the excited state is small, by varying such weight one can infer how the excited state would evolve alone (free of the ground state). This idea is somewhat trivial, but when applied to the time-dependent coupled cluster wave function, we obtain a set of equations that describe how the excited state, or a combination of quantum states, changes as a function of time. Link. Update: A follow-up of this work is available at the Electronic Structure journal, Link. This shows a generalization of the formalism and an improved description of populations and coherences.
Raman Signals and Electronic Features of 2D Heterostructures
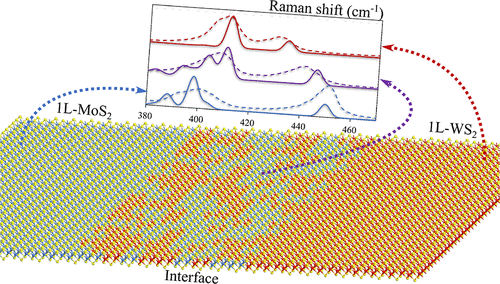
In this paper published last year at JPCC, we teamed up with Pierce Fix, Nicholas Borys (MSU Physics), and Patrick Kung (U. Alabama), to show that theory and experiment work together to quantify and interpret spatial non-resonant Raman spectra of 2D MoxW1-xS2. We track the evolution of the dominant modes as a function of composition in the heterostructure, and this includes the characterization of S-vacancy states. This allowed us to reverse-engineer the interface and observe that the averaged variation of composition in terms of position, indicating a smooth transition between the non-hybridized regions. Link
Welcome 2023
It has been a long time since the last post in this blog. The other pages have been updated though. 2023 is going to be different. New posts coming.
Size-Consistency and the Exponential Ansatz
We recently showed that the exponential Ansatz applied to DFT calculations eliminates fractional charges in spin-unrestricted calculations. It also offers ways to study, within single-particle formalisms, states where multiple electrons are excited (not just a single electron as in standard linear response methods). Looking forward to a more extended implementation of the method. This work is also compatible with the addition of two-body interactions. More info (Journal of Physical Chemistry A link): bit.ly/3oGgYq1

No C++ module support in G++ 10
After spending a good amount of time compiling gcc 10.2, I assumed the new, modules syntax was going to be enabled. Wrong, it’s not. Most new C++20 features are supported in v 10, except modules. Modules will be a big addition to C++, this link talks about it.
Hello world, again!
I am refreshing this blog
Second linear response code
The code that I’ve been working on for a while to compute excited state absorption spectra can be found here https://gitlab.com/plusxmartin/nwproj , the wiki page contains more details. This is work in progress. For more info please contact me
Germanium Fluorides
Our recent work on Germanium Fluoride nanocages was accepted by the Journal of the American Chemical Society. In this work, we studied with DFT a series of high-symmetry molecules with Formula GenFn, where n is between six and sixty. The molecules we studied show optical transparency or semi-transparency, electron delocalization, LUMO energies below -4 eV, and gaps in the range 1.6-3.2 eV. Although the formation energies are high, the fullerene-like systems have isomers with lower ground-state energies, where some Fluorine atoms are located inside the cages. So the fullerene below could undergo a transformation with F atoms preferring to be inside; but the barrier for such a transition should be very high. Link https://pubs.acs.org/doi/10.1021/jacs.8b11259